Javier e Illán: casos únicos en Asturias y raros en el mundo
Día de las enfermedades raras. Uno sufre un trastorno Nedamss. El otro, síndrome de Dent. Ellos lo llevan «con tremenda fortaleza». Y sus padres luchan para que se invierta más en investigación

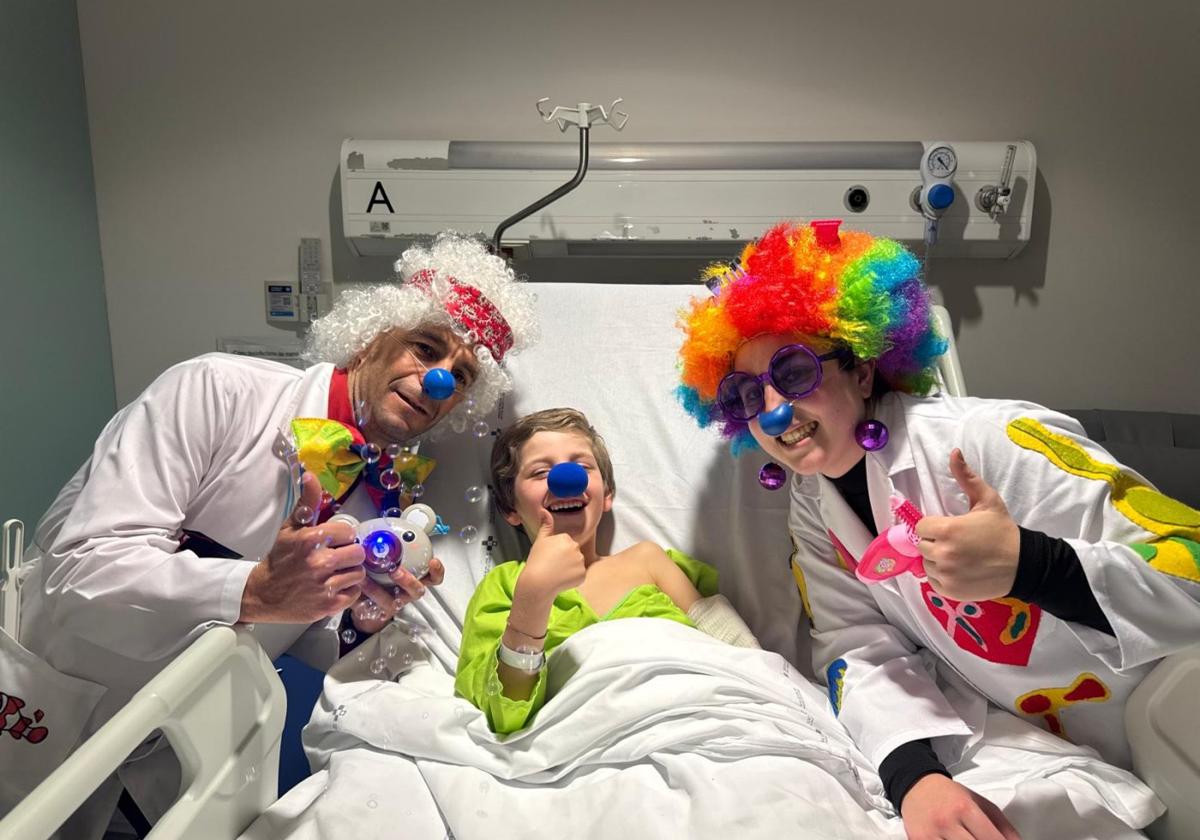
Hace unos días, Ana Piera recibía, de camino al Hospital Universitario Central de Asturias (HUCA), donde permanece ingresado su hijo pequeño, una llamada que ha cambiado radicalmente la vida de toda su familia. Al otro lado del teléfono, le informaban de que Javier, que está a punto de cumplir 8 años, padece el síndrome relacionado con el IRF2PBL, mutación genética que provoca un trastorno del neurodesarrollo con regresión, movimientos anormales, pérdida del habla y convulsiones (Nedamss). Una enfermedad ultra rara de la que, hasta este momento, sólo se habían diagnosticado 73 casos en el mundo y ocho en España. Este niño rubio, de ojos inmensos y una sonrisa «que ilumina a todos los que le rodean», es el noveno de nuestro país.
Hasta llegar a esta noticia tan brutal, ante la que resulta imposible contener las lágrimas, Ana Piera ha hecho lo imposible para encontrar explicación a los síntomas que Javier comenzó a manifestar hace justo un año. Un niño sano, al que le encantaba andar en bicicleta y jugar al fútbol, comenzó de repente a caminar de puntillas, a caerse con frecuencia y a desviar los ojos. Pero en las pruebas que le realizaron inicialmente no se apreciaba ninguna anomalía y el neuropediatra encargado de su caso concluyó que se trataba de un episodio de estrés que requería supervisión de los servicios psiquiátricos.

Detrás de éste, vinieron otros diagnósticos erróneos y algunos episodios que no perdona, todos ellos relacionados con el primer especialista que trató a su hijo. En su opinión, debería haber profundizado más en el caso de Javier, porque resultaba evidente que algo extraño le estaba ocurriendo. «Pasamos un calvario. Y lo que nos queda», comenta Ana Piera, que, ante el empeoramiento de su hijo, insistió una y otra vez en la necesidad de hacerle más pruebas genéticas. Tenía toda la razón. Lo que nunca imaginó es que el resultado de esos análisis iba a «dejarnos rotos».
Ahora, el futuro de Javier es incierto. El síndrome IRF2PBL o trastorno de Nedamss no tiene cura y provoca la pérdida progresiva de la capacidad de moverse, hablar o incluso tragar. Algo así como una ELA, pero en niños. «Es horrible», señala Ana Piera, que, siendo médica de Familia en el centro de salud Naranco y profesora asociada de la Facultad de Medicina, no es capaz de encontrar consuelo ni respuestas en la ciencia, porque, a día de hoy, poco se sabe de la enfermedad que sufre su hijo y su pronóstico «es infausto»
Según Ana Piera, «necesitamos un milagro», por el que no va a esperar con los brazos cruzados: «Hay que trabajar para que ocurra». Y ese trabajo le concierne a todo el mundo, porque «esto le puede pasar a cualquiera». Pero muy especialmente le concierne a las instituciones públicas, que «no pueden mirar para otro lado y tienen que invertir más en investigación».
En este momento, a Javier le cuesta ya mantenerse en pie, a pesar de «su tremendísima fortaleza». El HUCA está agilizando los trámites para su derivación al Hospital Sant Joan de Déu (Barcelona), a ver si allí encuentran alguna alternativa terapéutica que permita detener la «cruel» evolución de esta enfermedad neurodegenerativa. Mientras, la familia de Javier llora a gritos, con la esperanza de que se les escuche.
«Por favor, necesitamos que se investigue», clama Ana Piera, al tiempo que pone en valor el esfuerzo realizado por la asociación española de IRF2BPL para recaudar fondos. También Pedro Casares hace lo imposible para dar visibilidad a la enfermedad rara que padece su hijo Illán y poner el foco en «la falta de financiación» y en «la no investigación por parte de la industria farmacéutica». Al ser pocos los afectados, las patologías de baja prevalencia «no son rentables».
Illán tiene la enfermedad de Dent, un trastorno de carácter hereditario y origen genético que afecta a los riñones. Actualmente, sólo hay 240 casos diagnosticados en todo el mundo, 40 en España. El suyo –como le ocurre a Javier con el síndrome de IRF2BPL– es, además, el único caso detectado en Asturias. Cuando sus padres recibieron el diagnóstico, teniendo él 4 años, «lo pasamos fatal. Nunca habíamos oido hablar de esta enfermedad y no sabíamos qué hacer. Teníamos muchas preguntas y ninguna respuesta», explica Pedro Casares.
Ahora, Illán tiene 13 años y estudia 2º de la ESO. Toma cinco pastillas y media al día; bebe un mínimo de 5 litros de agua; y lleva una alimentación restrictiva en calcio y proteínas. Que sus riñones no funcionen correctamente altera, por ejemplo, la asimilación de nutrientes o el crecimiento de sus huesos. De hecho, recibe un tratamiento a base de hormonas del crecimiento que «el HUCA nos ha luchado» y debe someterse a controles periódicos: «Nos tratan de cine en Nefrología».
Hoy por hoy, el Dent no tiene cura y los fármacos disponibles «palian sólo los síntomas y están pensados para adultos». Aún así, Illán «hace una vida bastante normal», practica judo y toca el tambor en la Agrupación de San Salvador. Sus padres le han preparado para convivir con la enfermedad, que «seguramente acabará en trasplante». Quizá por eso asume con entereza de adulto todos los trastornos que conlleva. Y a través de la asociación Art for Dent buscan en la solidaridad privada la ayuda que no acaban de encontrar en las instituciones.
¿Tienes una suscripción? Inicia sesión